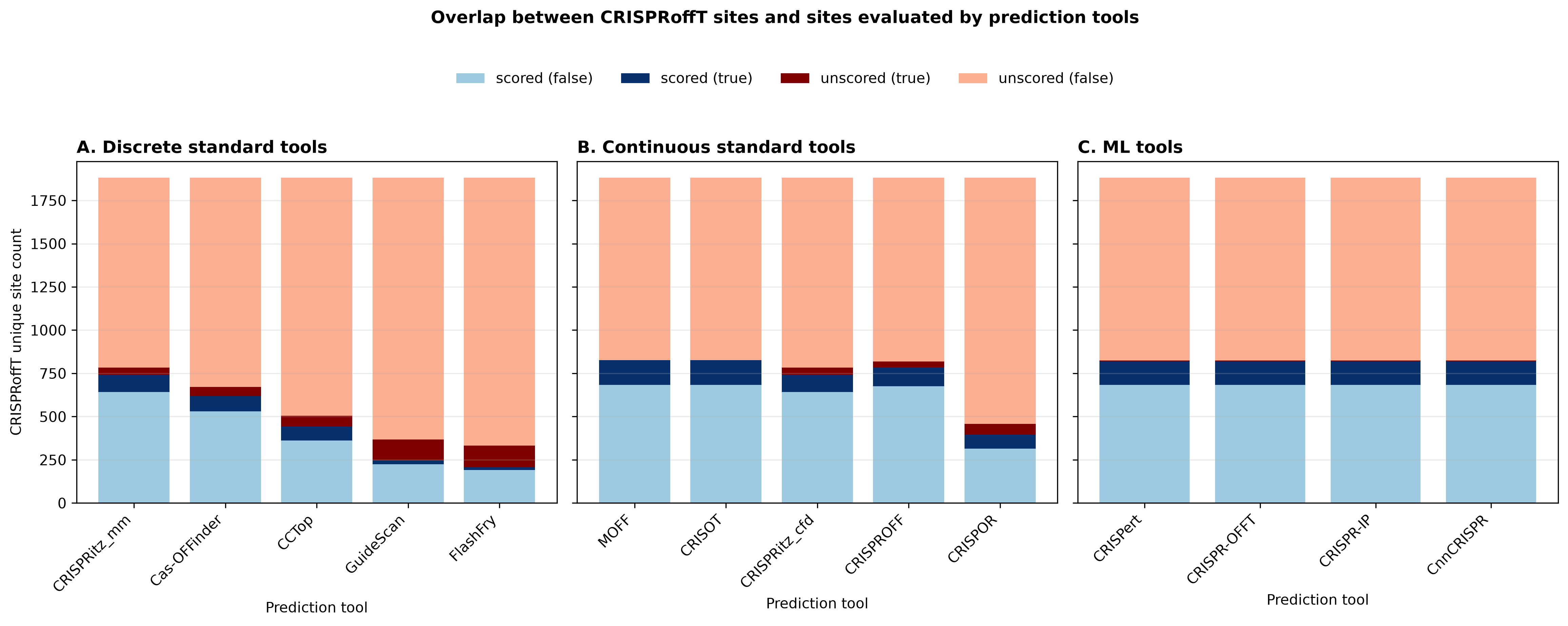
Figure 5 compares the CRISPRoffT off-target sites with the site sets evaluated by each prediction tool. The y-axis gives counts of unique CRISPRoffT site identities, defined by guide sequence, off-target sequence, and chromosome.
Each bar is split into four categories: validated true sites that were scored by the tool, labeled false sites that were scored by the tool, validated true sites that were not scored, and labeled false sites that were not scored. The panels separate discrete standard tools, continuous standard tools, and ML-based tools.
The ML tools were available only for the full-length no-bulge compatible candidate universe. In panel C, CRISPRoffT sites outside that compatible space therefore remain in the denominator and appear as unscored for the ML tools.
Inputs
The CRISPRoffT site set comes from the filtered manuscript truth table:
data/manuscript/manuscript_primary.csv
The standard prediction contracts are expected under:
data/zenodo/standard_tool_predictions/
The ML prediction contracts are expected under:
data/zenodo/no_bulge_ml_tool_predictions/
These prediction contracts are large Zenodo-backed artifacts. During local development they can be symlinked into the expected directories.
Load CRISPRoffT Site Identities
Code
import matplotlib.pyplot as pltimport numpy as npimport pandas as pdfrom offtarget_benchmark.layout import repo_layoutlayout = repo_layout()truth_path = layout.data_dir /"manuscript"/"manuscript_primary.csv"standard_prediction_dir = layout.data_dir /"zenodo"/"standard_tool_predictions"ml_prediction_dir = layout.data_dir /"zenodo"/"no_bulge_ml_tool_predictions"figure_dir = layout.docs_dir /"generated_figures"figure_dir.mkdir(parents=True, exist_ok=True)def normalize_truth_status(value):if pd.isna(value):return"unknown"ifisinstance(value, (bool, np.bool_)):return"true"ifbool(value) else"false" text =str(value).strip().lower()if text in {"true", "1", "yes", "off"}:return"true"if text in {"false", "0", "no", "on"}:return"false"return"unknown"truth = pd.read_csv(truth_path, low_memory=False)truth["truth_category"] = truth["truth_status"].map(normalize_truth_status)truth_sites = truth[truth["truth_category"].isin(["true", "false"])].copy()truth_sites["match_key"] = ( truth_sites["guide_seq"].astype(str)+"::"+ truth_sites["offtarget_seq"].astype(str)+"::"+ truth_sites["chromosome"].astype(str))truth_sites = truth_sites[["match_key", "truth_category"]].drop_duplicates("match_key").reset_index(drop=True)truth_sites["truth_category"].value_counts().rename_axis("truth_category").reset_index(name="n_unique_sites")
truth_category
n_unique_sites
0
false
1740
1
true
142
The figure uses unique site identities. This avoids counting the same site/tool overlap more than once when the source table contains repeated rows for the same guide, off-target sequence, and chromosome.
A site is scored when it appears in the standardized prediction contract for the tool. For native search tools this means the site was returned by the tool. For pair-scoring tools and ML tools this means the site was part of the candidate universe supplied to that scorer.