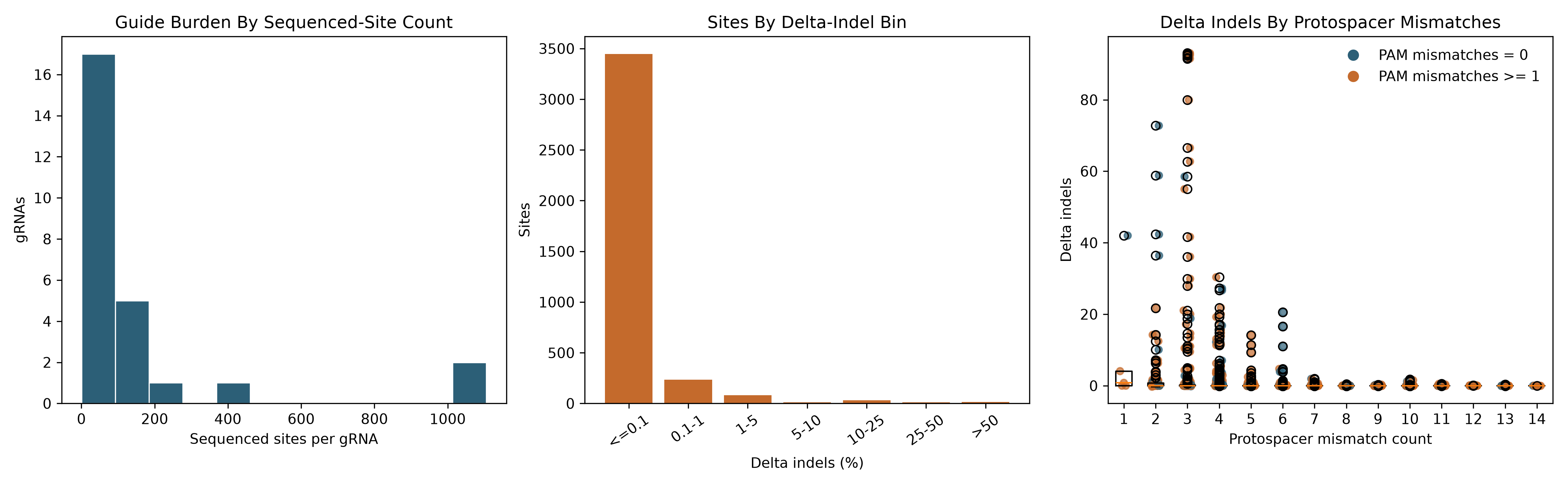
figure_dir = layout.docs_dir / 'generated_figures'
figure_dir.mkdir(parents=True, exist_ok=True)
fig, axes = plt.subplots(1, 3, figsize=(15.5, 4.8))
site_counts = sites_per_guide['n_sites'].dropna().astype(int).to_numpy()
n_bins = min(20, max(8, int(np.ceil(np.sqrt(len(site_counts)))) * 2)) if len(site_counts) else 8
axes[0].hist(site_counts, bins=n_bins, color='#2c5f77', edgecolor='white', linewidth=0.8)
axes[0].set_title('Guide Burden By Sequenced-Site Count')
axes[0].set_xlabel('Sequenced sites per gRNA')
axes[0].set_ylabel('gRNAs')
indel_bins = [-np.inf, 0.1, 1.0, 5.0, 10.0, 25.0, 50.0, np.inf]
indel_labels = ['<=0.1', '0.1-1', '1-5', '5-10', '10-25', '25-50', '>50']
binned = pd.cut(quantified['delta_indels'].astype(float), bins=indel_bins, labels=indel_labels, include_lowest=True)
indel_counts = binned.value_counts(sort=False).reindex(indel_labels, fill_value=0)
axes[1].bar(indel_labels, indel_counts.astype(int), color='#c46a2c', width=0.8)
axes[1].set_title('Sites By Delta-Indel Bin')
axes[1].set_xlabel('Delta indels (%)')
axes[1].set_ylabel('Sites')
axes[1].tick_params(axis='x', rotation=35)
categories = sorted(x for x in mismatch_df['protospacer_mismatch_count'].dropna().unique().tolist())
data = [
mismatch_df.loc[mismatch_df['protospacer_mismatch_count'] == cat, 'delta_indels'].astype(float).tolist()
for cat in categories
]
if data:
from matplotlib.lines import Line2D
axes[2].boxplot(data, tick_labels=[str(int(cat)) for cat in categories])
for idx, cat in enumerate(categories, start=1):
sub = mismatch_df[mismatch_df['protospacer_mismatch_count'] == cat]
colors = ['#2c5f77' if int(pam) == 0 else '#c46a2c' for pam in sub['pam_mismatch_count'].fillna(0)]
jitter = np.linspace(-0.12, 0.12, num=len(sub)) if len(sub) else []
axes[2].scatter(np.full(len(sub), idx) + jitter, sub['delta_indels'], s=24, alpha=0.7, c=colors)
legend_handles = [
Line2D([0], [0], marker='o', color='none', markerfacecolor='#2c5f77', markeredgecolor='#2c5f77', markersize=7, label='PAM mismatches = 0'),
Line2D([0], [0], marker='o', color='none', markerfacecolor='#c46a2c', markeredgecolor='#c46a2c', markersize=7, label='PAM mismatches >= 1'),
]
axes[2].legend(handles=legend_handles, loc='best', frameon=False)
axes[2].set_title('Delta Indels By Protospacer Mismatches')
axes[2].set_xlabel('Protospacer mismatch count')
axes[2].set_ylabel('Delta indels')
fig.tight_layout()
out_path = figure_dir / 'figure_1_dataset_overview.png'
fig.savefig(out_path, dpi=300)
plt.close(fig)
display(Image(filename=str(out_path)))